
Thyroid hormone – an iodine-containing molecule secreted by the thyroid gland – regulates development, growth, energy homeostasis, cardiovascular and muscle-skeleton systems, and cognitive function. Thyroid hormone is secreted into the circulation mostly in the form of a prohormone, T4. Much less (about 1/10) is secreted as T3, the biologically active form of thyroid hormone.
T4 and T3 enter virtually all cells through membrane transporters. Once inside the cells, T3 diffuses to the nucleus and interacts with thyroid hormone receptors (TRa or TRb) to modulate gene expression. T3-TR complexes control specific sets of T3-responsive genes, thus promoting T3-dependent biological effects. Tissues contain different combinations of TRa and TRb as well as other co-regulators of the T3-TR complex. Therefore, the effects of T3 depend on these combinations and are of course highly specific, individualized for each cell type. For example, T3 acts in the myocardium to accelerate heart rate and contraction force while at the same time T3 acts in the liver decreasing serum levels of cholesterol and other lipids.
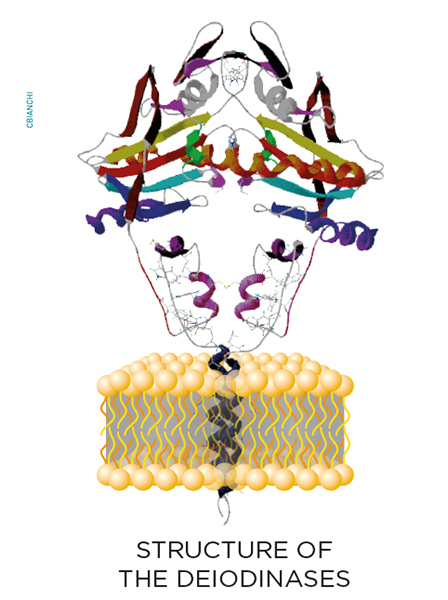
 While inside the cells, thyroid hormone molecules can be modified on a cell-specific fashion through the deiodinase group of enzymes. These are homodimeric Type I integral membrane selenoproteins composed of a single N-terminal trans-membrane segment connected to a larger globular domain with a Selenocysteine-containing active center embedded in a thioredoxin-like fold. Deiodinases modify the biological activity of thyroid hormone molecules either activating T4 (type II deiodinase, D2) or inactivating T4 and T3 (type III deiodinase, D3).
While inside the cells, thyroid hormone molecules can be modified on a cell-specific fashion through the deiodinase group of enzymes. These are homodimeric Type I integral membrane selenoproteins composed of a single N-terminal trans-membrane segment connected to a larger globular domain with a Selenocysteine-containing active center embedded in a thioredoxin-like fold. Deiodinases modify the biological activity of thyroid hormone molecules either activating T4 (type II deiodinase, D2) or inactivating T4 and T3 (type III deiodinase, D3).
As a result of the deiodinases, the flow of T3 molecules diffusing from the cell membrane to the nucleus can be increased with additional T3 supplied by the D2 pathway, which locally converts T4 to T3. In contrast, the D3 pathway decreases the flow of T3 to the nucleus because it terminally inactivates T3 to T2.
D2 is a short-lived ER-resident protein that generates T3 in a cell compartment that is adjacent to the nucleus. This probably explains why D2 activity results in higher TR occupancy with locally generated T3. In contrast, D3 sorts to the plasma membrane, where it undergoes endocytosys and recycling via early endosomes. Notably, D3 is also found incorporated into neurosecretory vesicles in hypothalamic neurons. Under hypoxic and/or ischemic conditions, D3 is redirected from the ER to the nuclear envelope, where it inactivates T3 and slows down cellular metabolism. The D1 pathway is not thought to affect intracellular thyroid hormone signaling other than by minimally contributing to the pool of circulating T3.
The expression of D2 and D3 can change rapidly in response to a number of developmental, metabolic, and disease cues through different signaling pathways. Because the expression of these enzymes can be turned on or off in discrete groups of cells, most of the time their actions do not affect circulating thyroid hormone levels, which are tightly controlled via the TRH/TSH/Thyroid axis. Thus, the actions of D2 and D3 are viewed as a cell-specific pre-receptor mechanism to control thyroid hormone signaling that cannot be predicted based on the levels of circulating thyroid hormone. For example, stimulation of D2 expression in brown adipose tissue accelerates transcription of T3-responsive genes such as UCP-1 and PGC-1, without elevating serum T3 levels. In contrast, ectopic D3 expression in the heart and brain during ischemia or hypoxia lifts the T3-dependent transcriptional footprint in these organs, in what can be seen as an adaptive mechanism to the disease state.